
BIP 2024 N°4

EDITORIAL
Desinformation sur le médicament : un sujet politique ?
La dernière élection américaine a illustré une fois de plus l’essor inquiétant de la désinformation. En matière de santé, la nomination de l’ancien démocrate Robert F. Kennedy Jr à la tête de la santé des États-Unis, donc en charge de la réglementation des médicaments, est inquiétante. Parmi d’autres relais de fake news, l’avocat et neveu de JFK s’est fait connaître pour ses positions antivaccins, remettant publiquement en cause leur efficacité et alimentant les rumeurs sur un lien entre vaccination et autisme. Trump a promis qu’il le laisserait « se déchaîner sur les médicaments », un programme inquiétant.
En France, la classe politique n’est pas non plus toujours au rendez-vous pour lutter contre la désinformation sur les médicaments. Un exemple marquant est celui de Patrick Hetzel, dernier ministre de l’Enseignement supérieur et de la Recherche, dont les prises de position sur les médicaments sont pour le moins discutables. Selon le journal Libération, il avait publiquement défendu l’usage de l’hydroxychloroquine promue par Didier Raoult dans la COVID-19. Pour lui, nous avions « la preuve empirique de son efficacité », une conception singulière de la démarche scientifique. En 2021, il s’était également opposé à l’obligation vaccinale et avait relayé la rumeur selon laquelle le Rivotril® aurait été largement utilisé pour euthanasier des patients. En 2018, l’actuel ministre avait déposé un amendement destiné à ralentir le déremboursement de l’homéopathie, en soutien à une filière qu’il considérait « lourdement fragilisée par des années de dénigrement ». Le texte avançait que « de nombreuses études ont démontrés (sic) que les patients pris en charge par des médecins homéopathes ont un bénéfice clinique équivalent qu’avec une prescription de médicaments classiques ». Une affirmation trompeuse et, encore une fois, sans fondement scientifique.
Face à la montée inquiétante de la désinformation en santé, il est important de rappeler la responsabilité de nos dirigeants. Alors qu’auprès des patients la communication d’informations validées sur le bon usage du médicament est de plus en plus difficile, il est malheureux de devoir espérer que ces hommes politiques éloignés de la science ne fassent pas trop de dégâts...
Jean-Luc Faillie, Haleh Bagheri, Agnès Sommet
Sommaire
- INFOS IMPORTANTES - ALERTES
- PHARMACOLOGIE CLINIQUE
- PHARMACOVIGILANCE
- Dispositifs transdermiques et IRM : risque de brûlure
- Le syndrome 3 M immuno-induit ou syndrome de chevauchement myocardite, myosite, myasthénie au décours d’une immunothérapie anticancéreuse par inhibiteur de point de contrôle immunologique
- Nicorandil et ulcérations cutanéo-muqueuses : Série Netflix en une dizaine d’épisodes !
- Erreurs liées au méthotrexate par voie orale : un problème persistant malgré les mesures
- PHARMACO-EPIDEMIOLOGIE
- MEDICAMENTS, REPRODUCTION ET PERINATALITE
- PHARMACOLOGIE SOCIALE
- PHARMACOLOGIE-TOXICOLOGIE
A LA UNE !
Vasoconstricteurs à base de pseudoéphédrine par voie orale
Quel choc ce matin du 10 décembre 2024 en écoutant tranquillement la radio : entre les informations sur la chute du régime syrien et le choix du futur premier ministre, la journaliste explique que l’ANSM annonce qu’une ordonnance devra obligatoirement être présentée par un patient pour obtenir les vasoconsctricteurs (à base de pseudoéphédrine par voie orale) en pharmacie !!! Suis-je réveillée ? Est-ce un rêve qui devient réalité ?
« Tout vient à point pour qui sait attendre »
Ce proverbe pourrait s’appliquer à la « saga » des vasoconstricteurs décongestionnants de la sphère ORL. L’autre question qui émerge serait « mais fallait-il attendre autant de temps pour prendre cette décision d’obligation de prescription » ? Bien sûr que non. La proposition d’obligation de prescription était pourtant énoncée et argumentée depuis 20 ans (oui vous lisez bien, 20 ans) par le réseau des CRPV et plus particulièrement par le CRPV de Toulouse, chargé des différentes enquêtes de pharmacovigilance (8 rapports). Depuis les premières alertes en 2001, 2 directeurs du CRPV et 3 experts se sont succédés au CRPV de Toulouse, argumentant sans relâche sur les risques en lien avec les propriétés pharmacologiques de ces médicaments, leur risque de mésusage et leur bénéfice clinique modeste voir absent. Les données prouvant l’absence de rapport bénéfice/risque favorable de ces médicaments étaient donc pourtant disponibles depuis longtemps. Que fallait-il de plus pour prendre des décisions plus rapides et ainsi rendre « évitables » les effets indésirables, certes rares, mais graves (AVC, IdM, poussées hypertensives,…) survenus ces dernières années ? Vaste question qui appelle quelques réflexions:
le temps d’analyse des données par les experts ne correspond pas au temps de communication des décisions par les autorités, avec des allers-retours incessants entre l’EMA et l’ANSM
même pour une pathologie bénigne (le rhume) traitée par des médicaments à risque, la décision de prescription obligatoire semble difficile à prendre !
parmi les qualités requises pour un expert en PV, on peut citer la ténacité, la patience et la persévérance !
Oui de la patience, enfin récompensée par cette décision salutaire pour les patients, dont les symptômes du rhume s’amélioreront en 1 semaine sans médicaments à défaut de 7 jours sous vasoconstricteurs !!!
Point d'information de l'ANSM : Actualité - Rhume : ordonnance obligatoire pour toute dispensation de médicament à base de pseudoéphédrine - ANSM
Pascale Olivier (Toulouse)

L'ordonnance du mois
François Montastruc (Pharmacologie Clinique, Toulouse) et Dr Rémy Klein (Psychiatre, Toulouse)
Vous voyez en consultation Madame A. 35 ans pour la première prescription d’un traitement médicamenteux par lithium pour la prise en charge d’un trouble bipolaire. La patiente est suivie en cardiologie pour une hypertension artérielle traitée par une association ramipril 5mg et hydrochlorothiazide 12,5mg. En plus de ces médicaments, elle prend aussi de l’ibuprofène 400 mg, 1 à 2 comprimés par jour pour ses crises migraineuses régulières.
Questions :
- Concernant la prescription de lithium, quelles sont les contre-indications que vous devez rechercher chez cette patiente ?
- Le lithium présente un profil de risque important avec beaucoup d’effets indésirables potentiels. Selon vous, de quels effets indésirables devez-vous prévenir en priorité votre patiente avant de prescrire le lithium ?
- Que pensez-vous de l’utilisation de l’ibuprofène chez cette patiente ?
Rappel Pharmacologique
Le lithium est un ion Li+ indiqué dans les troubles bipolaires dont le mécanisme d’action n’est pas clairement établi (diminution de l’activité dopaminergique et/ou glutamatergique, action pro-sérotoninergique). C’est un médicament à marge thérapeutique étroite (la dose minimale au-dessous de laquelle le médicament est le plus souvent inactif et la dose maximale au-dessus de laquelle apparaissent le plus souvent des effets indésirables dose dépendants sont proches) nécessitant un suivi thérapeutique pharmacologique (dosage de la lithiémie). Son élimination est essentiellement rénale.
Le ramipril est un inhibiteur de l'enzyme de conversion diminuant l’activité du système rénine-angiotensine-aldostérone.
L’hydrochlorothiazide est un diurétique thiazidique. Les diurétiques thiazidiques agissent par blocage du cotransport Na+/Cl- au niveau du segment de dilution (segment proximal du tube contourné distal).
L’ibuprofène est un AINS. Par leur action inhibitrice de la COX, tous les AINS inhibent la transformation de l’acide arachidonique en prostaglandines et thromboxanes.
Réponses :
Contre-indications au lithium : hypersensibilité ; insuffisance rénale en cas d’impossibilité d’exercer une surveillance stricte de la lithiémie et la fonction rénale ; insuffisance cardiaque ; maladie d’Addison ; syndrome de Brugada ou antécédent familial. L’allaitement est aussi une contre-indication stricte car le lithium passe largement dans le lait maternel (50% de la concentration plasmatique). Dans ce cas nous vous proposons de contacter les équipes de pharmacovigilance « médicaments et allaitement » pour discuter la balance bénéfices/risques du lithium lors de l’allaitement.
Information à donner en priorité concernant les effets indésirables. Le lithium est associé à de nombreux effets indésirables. Lors d’une première prescription, il paraît difficile de lister de façon exhaustive les effets indésirables. Les patients doivent cependant être informés de certains effets indésirables potentiellement « graves » et qui vont motiver les examens cliniques et paracliniques avant la prescription. Nous proposons de retenir ces effets indésirables en fonction des 5 systèmes : Rein, Parathyroïde/thyroïde, Neurologie, Coeur et Peau :
Rein - Le lithium peut être à l’origine à moyen terme de syndrome polyuro/polydypsique, de diabète insipide néphrogénique et de syndrome néphrotique. La surveillance de la fonction rénale (protéinurie) est donc clé lors de la prise de lithium à la fois pour l’adaptation de la lithiémie mais aussi sur la surveillance des effets indésirables rénaux. Sur des expositions prolongées, le lithium peut-être aussi responsable d’insuffisance rénale par néphropathie tubulo-interstitielle chronique.
Thyroïde/parathyroïde - Le lithium altère la fonction parathyroïdienne et le métabolisme de calcium. On estime que 40% des patients sous lithium pourraient développer une hypercalcémie souvent associée à une hyperparathyroïdie. Le lithium est aussi à l’origine d’hypothyroïdie (20%) ou d’hyperthyroïdie (très rare).
Neurologie - Parmi les effets indésirables les plus fréquents, on retrouve les tremblements des extrémités et/ou des plaintes cognitives, troubles de la mémoire, une sédation qui peuvent-être les premiers signes cliniques d’un surdosage en lithium. Les complications plus « graves » sont des tableaux de confusion, états convulsifs (EEG à faire si antécédent), rarement syndrome sérotoninergique (en association IRS, tramadol), encéphalopathie et syndrome cérébelleux (habituellement réversible).
Cœur - Un examen cardiovasculaire et un ECG sont nécessaire avant l’initiation du lithium en raison du risque potentiel de troubles du rythme (bradycardie sévère, bloc sino-auriculaire, allongement du QTc)
Peau - L’acné ou les éruptions acnéiformes sont des atteintes méconnus avec lithium mais qui pourrait toucher jusqu’à 30% des patients exposés. Le lithium provoque aussi des aggravations ou des déclenchements de psoriasis ou d'éruptions psoriasiformes, alopécie. Le lithium est à l’origine d’autres effets indésirables comme des effets digestifs (diarrhée et nausées) une prise de poids, une hyperleucocytose, ou des effets oculaires, œdèmes…. Dans ce cas clinique, il ne faudra pas oublier de faire un test de grossesse avant de prescrire le lithium.
Attention aux AINS – ibuprofène ! Avant d’évoquer l’interaction pharmacocinétique (sur l’élimination rénale) entre la prise d’AINS (mais aussi diurétiques et IEC) et le lithium, il faut rappeler le « réflexe de pharmacovigilance » devant tout symptôme chez tous les patients : “Et si c’était le médicament ? ” !!!!
Il faut donc d’abord s’interroger sur l’origine de son hypertension artérielle et questionner la responsabilité de la prise régulière d’ibuprofène. En effet, tous les AINS augmentent la pression sanguine artérielle jusqu’à 10 mmHg. Cette action s’explique par l’augmentation de la volémie et la vasoconstriction systémique induite par l’inhibition de l’action des prostaglandines. Une (ré)évaluation de la pertinence clinique de l’usage régulier de l’ibuprofène est nécessaire (et discuter des alternatives). L’arrêt de l’ibuprofène doit permettre de revoir le traitement médicamenteux antihypertenseur.
Concernant l’interaction entre les AINS, les diurétiques, les IEC avec le lithium, elle expose la patiente à un risque de surdosage en lithium en cas d’aggravation de la fonction rénale. Les AINS d’abord et les diurétiques et IEC (ou ARA2) sont des facteurs médicamenteux à l’origine d’insuffisance rénale fonctionnelle et diminuant l’élimination urinaire de lithium (compétition sur la réabsorption de sodium). Il faudra donc informer la patiente de ces interactions et en particulier que la prise ponctuelle d’AINS expose à un risque de surdosage.
Utilisation hors AMM de l’ivermectine dans le traitement de la pédiculose pubienne sévère
Ariane OLIVAN (interne) et Pascale PALASSIN (Montpellier)
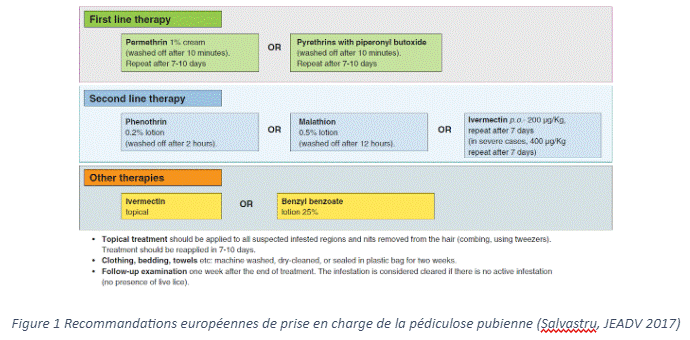
La pédiculose pubienne, communément appelée « morpions », est une parasitose transmise majoritairement par contact rapproché avec une personne infestée par Phthirus pubis, un parasite visible à l’œil nu, responsable de démangeaisons au niveau des zones pileuses infectées.
La mise en place de mesures d’hygiène rigoureuses est généralement suffisante pour éviter toute ré-infestation. Il est conseillé de décontaminer le linge et la literie, par un lavage à haute température (T°> à 50 °C) et d’aspirer la surface du matelas. Les articles non lavables doivent être mis dans des sacs en plastique scellés pendant 2 semaines. Le rasage des zones infestées permet d’éliminer les lentes non écloses. Le traitement des partenaires sexuels (sur les trois derniers mois) et le dépistage des autres infections sexuellement transmissibles (IST) est indispensable, 30 % des cas de pédiculose pubienne étant associés à une autre IST (1).
Si les démangeaisons sont importantes, l’application de produits anti-poux classiques est recommandée en première intention, comme la perméthrine (paralysant) ou de pyréthrines associées au butoxyde de pipéronyle (inhibition des enzymes de détoxification). Ces topiques sont à appliquer sur les zones pileuses infestées pendant 10 minutes, avant d’être rincés, à J0 et J8 pour éviter la ré-infestation due aux œufs éclos.
Si une éradication complète n’a pu être obtenue, un traitement de deuxième ligne peut être envisagé par ivermectine, phénothrine ou malathion (2). L'ivermectine, paralyse spécifiquement les parasites en perturbant la neurotransmission en agissant sur les canaux ioniques dépendant du glutamate.
Selon les recommandations françaises (2022) et européennes (2017), un traitement oral par ivermectine à une posologie de 200 à 400 µg/kg à J0 et J8, bien qu’hors AMM en France, a fait la preuve de son efficacité dans les infestations les plus sévères (grade C) (1,3). Ces recommandations sont notamment basées sur les résultats d’un essai clinique multicentrique randomisé en double aveugle, qui comparait l’efficacité de l’ivermectine orale à une dose de 400 µg/kg au malathion 0,5% en lotion dans les cas d’infestations sévères par les poux de tête (2).
3. https://epopi.fr/?page=fiche&id=366&cat_id=259&tk=66730d82444338dfb7cc51de52338866
Le lenacapavir, une avancée pour la prophylaxie préexposition (PrEP) ?
Pauline Schiro (Toulouse)
Le lénacapavir est un antirétroviral qui se lie à la capside du VIH-1, bloquant plusieurs étapes clés du cycle viral. Il s’agit du premier antirétroviral avec un mécanisme d’action multiple autorisé dans l’Union Européenne. En France, il est indiqué en dernier recours, en association avec un autre antirétroviral chez les adultes infectés par le VIH-1 multirésistant. En raison d’une longue demi-vie d’élimination (8 à 12 semaines), une seule injection sous-cutanée tous les 6 mois est recommandée.
Actuellement, en France, pour la prophylaxie pré-exposition (PrEP) du VIH, le seul médicament disponible est l’emtricitabine/ténofovir disoproxil, par voie orale en prise continue ou à la demande (seulement chez les hommes). Son efficacité est confirmée à condition que l’observance soit bonne. C’est pourquoi l’arrivée sur le marché de médicaments tels que le lénacapavir suscite beaucoup d’espoir, notamment pour les pays où l’accès aux soins est limité.
Le laboratoire Gilead, qui commercialise le lénacapavir, a récemment publié les résultats de l’essai PURPOSE 1 (1), visant à comparer le lénacapavir (injection semestrielle) et l’emtricitabine/ténofovir (prise quotidienne) dans la prévention de l’infection par le VIH chez les jeunes femmes cisgenres. Suite aux résultats favorables de l’analyse intermédiaire, l’essai a été interrompu prématurément. Effectivement, aucune infection n’est apparue dans le groupe lénacapavir à l’inverse du groupe témoin emtricitabine/ténofovir. Ces résultats s’expliquent par une mauvaise observance dans le groupe nécessitant une prise quotidienne, alors que celle-ci a été bonne dans le groupe lénacapavir.
Des éléments pharmacologiques sont cependant à prendre en compte: sa longue demi-vie rend compliquée la gestion du risque de résistances, mais également celle des effets indésirables qui peuvent persister après l’arrêt. De plus, le lénacapavir est substrat et inhibiteur du CYP34A. Par conséquent, de multiples interactions médicamenteuses sont à prévoir, même plusieurs mois après l’injection.
D’autres essais sont en cours afin d’évaluer l’efficacité du lénacapavir dans la PrEP chez d’autres populations (2). Pour l’instant, Gilead n’a pas encore soumis de demande de mise sur le marché pour la PrEP. Par ailleurs, un autre antirétroviral, le cabotégravir, vient d’avoir le feu vert au remboursement par la HAS pour la PrEP en injection bimensuelle.
Dispositifs transdermiques et IRM : risque de brûlure
Clémence Jutard (interne), Pascale Palassin (Montpellier)
La présence de dispositifs transdermiques (patchs) pendant une imagerie par résonance magnétique (IRM) expose à un risque de brûlure. Ce risque a été signalé en 2006 par l'AFSSAPS (1).
En 2014, l’ANSM a publié des documents visant à améliorer le bon usage des dispositifs transdermiques et avertir les patients et professionnels de santé des risques liés à leur utilisation (2). En effet, certains patchs contiennent au niveau de la couche externe un feuillet de protection métallique, souvent en aluminium. Bien que non ferromagnétique, l'aluminium est un métal conducteur et peut entraîner des brûlures sous l'effet des courants électriques produits par les champs de radiofréquences des IRM.
Ainsi, l'ANSM recommande le retrait du dispositif transdermique avant l’IRM et son remplacement à l’issue de l’examen, à moins d’être certain de l’absence de matériau conducteur. En cas de retrait du patch, les modalités d’interruption du traitement doivent être discutées avec le prescripteur en fonction du médicament concerné (3).
Les informations relatives à la présence de métaux conducteurs dans le patch peuvent être retrouvées dans le Résumé des Caractéristiques du Produit (RCP) et la notice du médicament. Il est important de noter que la présence d’aluminium peut varier selon le laboratoire pour une molécule donnée, et que celle-ci ne fait pas nécessairement l’objet d’une mise en gardevis-à-vis de l’IRM dans le RCP.
Il apparaît ainsi essentiel, avant la réalisation d’une IRM, d’interroger systématiquement les patients sur tous leurs médicaments, y compris ceux sous forme de patch, et de connaitre la spécialité exacte, afin de vérifier sa composition. A noter que ce risque a également été rapporté avec les pansements à l’argent.
Classe thérapeutique | Exemples de dispositifs transdermiques |
Substituts nicotiniques | Nicopatchlib®, Nicoretteskin®, nicotine, Nicotinell®, Niquitin® |
Antalgiques / anesthésiques | Durogesic®, fentanyl, Matrifen®, Emlapatch®, Qutenza®, Versatis® |
Traitements préventifs de la crise d’angor | Discotrine®, Epinitril®, Nitriderm®, Trinipatch®, trinitrine |
Contraceptifs | Evra® |
Traitements hormonaux substitutifs | Demestril®, Demestril septem®, Estrapatch®, Femsept®, Femseptcombi®, Femseptevo®, Thais®, Thaissept® |
Anticholinestérasiques | Exelon®, rivastigmine |
Anticholinergiques | Scopoderm® |
Agonistes dopaminergiques | Neupro® |
Références :
Le syndrome 3M immuno-induit ou syndrome de chevauchement myocardite, myosite, myasthénie au décours d’une immunothérapie anticancéreuse par inhibiteur de point de contrôle immunologique
Sika OKOUMASSOUN (étudiante en Pharmacie Montpellier), Joanna Lapalus (interne en pharmacie Toulouse), Romain Barus (Toulouse) et Pascale PALASSIN (Montpellier)
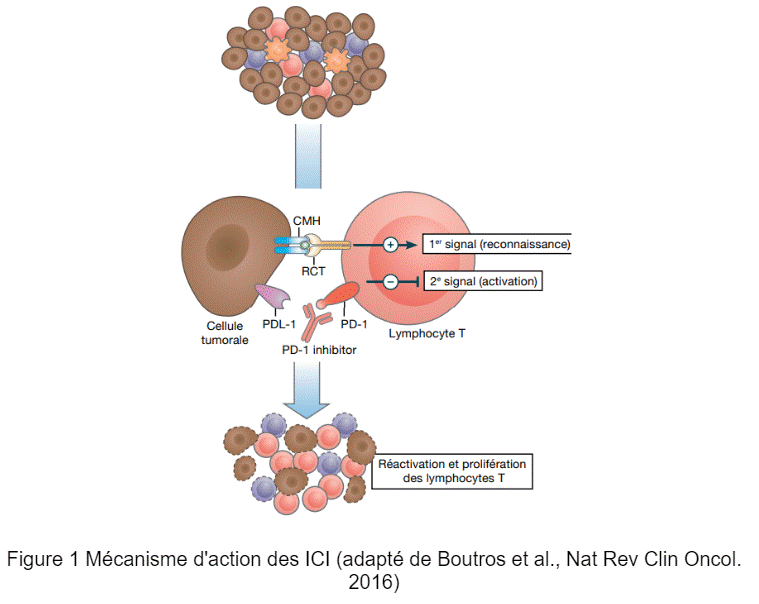
L’immunothérapie anticancéreuse par inhibiteurs de points de contrôle immunologique (ICI) a révolutionné la prise en charge de nombreux cancers. Elle se base sur la stimulation du système immunitaire pour neutraliser les cellules cancéreuses. Les ICI sont associés à un large spectre d’effets indésirables dits immuno-induits dont certains sont potentiellement fatals, comme le « syndrome 3M ».
Ce syndrome correspond au chevauchement de trois effets indésirables majeurs des ICI que sont la myocardite, la myosite et la myasthénie. Parmi les patients traités par ICI, environ 1% vont développer une myocardite immuno-induite, dont 25% vont développer simultanément une myosite et 11% un syndrome myasthénique associé (1).
Si ces 3 effets à médiation immunitaire ainsi que la conduite à tenir (arrêt du médicament et initiation de corticoïdes) sont bien décrits de façon indépendante dans les RCPs des ICI, le syndrome 3M n’est quant à lui pas mentionné stricto sensu. Parmi les anti-PD1 et anti-PD-L1, seul le cémiplimab (LIBTAYO®) mentionne dans sa monographie que la myosite, la myocardite ou la myasthénie grave peuvent survenir simultanément. Il est fait mention d’un « overlap » entre myosite et myocardite dans la monographie de l’atézolizumab (TECENTRIQ®), un anti PD-L1 et du nivolumab (OPDIVO®), un anti PD-1.
Une revue récente a décrit 50 patients atteints de syndrome 3M : la majorité étaient traités par ICI dans le cadre d’un mélanome (28%), d’un cancer pulmonaire (20%) ou d’un cancer rénal (14%). Le taux de mortalité à l’hôpital était de 38% (1). Les symptômes les plus fréquemment rapportés, devant faire suspecter un syndrome 3M, comprennent une ptose palpébrale (58%), une dyspnée (48%), une diplopie (42%) ou une myalgie (36%). Le délai moyen de survenue est d’environ 21 jours après la première cure. Les corticostéroïdes représentent le principal traitement de ces effets indésirables et doivent être administrés le plus précocement possible, sans attendre la confirmation diagnostique. Une recherche complémentaire dans la base internationale de pharmacovigilance de l’Organisation Mondiale de la Santé retrouvait 92 cas impliquant les anti-PD-1 ou anti-PD-L1 avec pour issue, le décès dans 37% des cas, concordant avec les données de la littérature.
En pratique, ce syndrome de chevauchement est une complication rare mais gravissime des ICI qui nécessite d’être identifiée rapidement pour en optimiser la prise en charge hospitalière en urgence. Les signes cliniques décrits plus hauts, associés à une élévation de la troponine ou des CPK doivent alerter les professionnels de santé sur ce risque potentiellement fatal.
En cas de question concernant la suspicion d’un effet indésirable immuno-induit, il est possible de faire appel aux Réunions de Concertation Pluridisciplinaires (RCP) régionales de recours, telle que la RCP Toxicités et Immuno-Oncologie en Occitanie-Est (ex-région Languedoc-Roussillon) et la RCP Immunotoxicités Occitanie-Ouest (ex-région Midi-Pyrénées), en inscrivant votre patient sur le Dossier Communicant de Cancérologie Onco-Occitanie afin d’obtenir un avis collégial concernant la prise en charge de ces effets indésirables mais aussi les possibilités éventuelles de reprise de l’immunothérapie.
Référence
Contacts RCP régionales sur la gestion des toxicités de l’immunothérapie :
- RCP Toxicités et Immuno-Oncologie (Montpellier) :
Coordonnée par le Dr. Alexandre Maria (médecin interniste) et le Pr. Xavier Quantin (onco-pneumologue)
1 réunion hebdomadaire tous les lundis de 16h à 18h
Pour accéder à la réunion en mode virtuel merci de contacter le secrétariat RCP au 04.67.33.55.29 ou secretariat-rcp@chu-montpellier.fr
- RCP Immunotoxicité (Toulouse) :
Coordonnée par le Dr. Valérian Rivet (médecin interniste) et le Dr. Iphigénie Korakis (oncologue médicale)
1 réunion mensuelle tous les derniers vendredis du mois
Inscription des patients via le DCC ou prise de contact avec le Dr. Rivet au 05 31 15 64 54 ou rivet.valerian@iuct-oncopole.fr
Nicorandil et ulcerations cutaneo-muqueuses : série Netflix en une dizaine d’épisodes !
(Haleh Bagheri-Toulouse)
Le nicorandil (Ikorel°, Adancor°), un activateur de canaux potassiques classé parmi les « Autres vasodilatateurs utilisés dans les maladies cardiaques », a obtenu son AMM en 1992 (et est génériqué depuis 2007) dans l’indication “traitement symptomatique des patients adultes atteints d’angor stable non contrôlé ou avec une contre-indication aux anti-angineux de première intention (bêta-bloquants ou inhibiteurs calciques)”. La notice du médicament indiquait le risque d’ulcérations au niveau gastro-intestinal ou oculaire. Une enquête de pharmacovigilance en 2012 avait analysé 170 cas d’ulcérations déclarés en France avec le nicorandil motivant une lettre aux prescripteurs le 20 mars 2012 pour les informer sur le risque d’ulcérations. Plusieurs séries de cas ont également été publiées dans la littérature (1).
L’hypothèse physiopathologique implique le métabolisme hépatique du nicorandil, donnant naissance à un dérivé inactif libérant de l’acide nicotinique et du nicotinamide qui intègrent le pool NAD/NADP du métabolisme cellulaire. En cas de surdosage ou suite à une augmentation de la posologie, l’acide nicotinique et le nicotinamide seraient amenés (par un mécanisme de saturation connu) à se distribuer en dehors de ce pool. Ils pénètreraient alors dans des zones lésionnelles préexistantes ou en phase de cicatrisation, provoquant ainsi une ulcération. Le nicotinamide (un ouvreur potassique) favoriserait par ailleurs la prolifération épithéliale des zones lésionnelles par augmentation du flux sanguin dans les tissus situés en bordure de la cicatrice. Cette hypothèse de lésion « chimique » est renforcée par les caractéristiques particulières de ces ulcérations : profondes, à bord net et fond propre, sans œdème, douloureuses, persistantes, s’aggravant progressivement, conduisant parfois à la formation de fistules et histologiquement non spécifiques (2).
Au niveau européen, des actions ont été menées par l’European Medicines Agency en 2013 et 2015 pour conclure en juin 2015 à un rapport bénéfices/risques toujours favorable mais nécessitant la réalisation en 2016 d’une étude observationnelle PASS (Post Authorization Safety Study) dont les résultats motiveront le rajout des effets indésirables liés à l’ulcération et leur fréquence dans la notice du produit en février 2021. En octobre 2022, une nouvelle revue des cas d’ulcérations n’a abouti qu’à une proposition d’un suivi étroit des cas (close monitoring).
En France, les chiffres de vente du nicorandil ont nettement baissé (de 4 000 000 boîtes en 2007 à 1 000 000 en 2023 à l’officine, même évolution à l’hôpital). A ce jour, le Service Médical Rendu (SMR) du nicorandil est toujours jugé important (avis HAS de 2015).
Les prochains épisodes se dérouleront au niveau européen, avec une réévaluation du médicament en mai 2025et au niveau français, avec la sollicitation de la HAS pour une réévaluation du SMR et une re-sensibilisation des professionnels de santé… Cependant, il est à noter que le laboratoire fabricant (Sanofi Winthrop) envisage d’arrêter la commercialisation du princeps Ikorel° en fin d’année 2024 ! Dommage, et alerte aux amateurs des séries Netflix, la saga «Nicorandil» va peut-être s’arrêter sur la décision du laboratoire fabricant. Enfin, on peut s’interroger sur le temps passé et le coût de toutes ces études inutiles avec la mise en évidence d’un risque il y a une douzaine d’années grâce à la notification spontanée.
1. Nicorandil and cutaneous ulcerations, their misdiagnosis and consequences : illustrations by five case reports and a review of the French pharmacovigilance database. Béné J, Carpentier O, Sabanowski S, Laroche ML, Beyens MN, Lambert M, et al. Therapie. 2018;73(5):409-417. doi: 10.1016/j.therap.2018.01.004. Epub 2018 Jan 31.
2. Nicorandil et ulcérations cutanée. Schmutz JL, Trechot P. Annales de dermatologie et de vénérologie, 2012, 139, 249-50.
Erreurs liées au méthotrexate par voie orale : un problème persistant malgré les mesures
Virginie Bres (Montpellier)
Le méthotrexate (MTX), utilisé dans le traitement de pathologies inflammatoires et oncologiques, est un médicament à marge thérapeutique étroite et il est associé à un risque important d'erreurs médicamenteuses, notamment de rythme d’administration. Ces erreurs consistent en une prise quotidienne de ce médicament, alors qu'elle doit être hebdomadaire dans le traitement des maladies inflammatoires. Un exemple récent illustre ces risques : un patient a confondu Imenor, une forme orale de MTX, avec un autre médicament, Tahor, et a pris simultanément Imenor et Imeth, une autre forme orale de MTX. Cette erreur a conduit à une aplasie médullaire, un effet indésirable grave du MTX. Cette erreur met en évidence un problème récurrent, soulignant la nécessité, au-delà de la vigilance, de mesures concrètes.
Les risques de toxicité liés à des erreurs médicamenteuses répétées par voie orale sont très préoccupants et figurent depuis 2012 sur la liste des « Never Events ». Des efforts ont été faits pour sensibiliser les patients et les professionnels de santé à ce problème. L'Agence européenne des médicaments (EMA) a réalisé une évaluation du risque d'erreurs de dosage du MTX en 2019, aboutissant à la mise en place de plusieurs mesures préventives, dont des supports éducatifs et des modifications de l'information produit. Parmi les propositions, la réduction du nombre de comprimés par boîte n'a toutefois pas été retenue. Malheureusement, les résultats de l’étude d'impact réalisée en 2022 dans cinq pays européens ont montré que les mesures mises en place entre 2019 et 2021 n'ont pas été suffisamment efficaces pour prévenir les erreurs médicamenteuses liées à l'utilisation du MTX (1).
Dans l'attente de nouvelles mesures, il est important pour les professionnels de santé de veiller à une communication claire avec les patients concernant les traitements par MTX, notamment lors des changements de prescription. Il convient également de s'assurer que le patient comprend bien les instructions et l'importance de suivre strictement la posologie prescrite.
Risque de troubles du neurodeveloppement chez les enfants de pères exposes au valproate : quelles données ?
Isabelle Lacroix (Toulouse)
Lors d’exposition pendant la grossesse au valproate, les risques d’anomalies congénitales et de troubles du neurodéveloppement de l’enfant sont bien connus. En 2018, dans le cadre d’une réévaluation des risques liés à ce médicament, l’Agence Européenne du Médicament (EMA) a demandé aux firmes le commercialisant de fournir des données sur les risques potentiels liés cette fois à une exposition paternelle. Une étude (non publiée) a été réalisée à partir de plusieurs registres de naissances scandinaves (Norvège, Suède et Danemark) : elle a comparé 2121 enfants de pères traités par valproate dans les trois mois qui précédaient la conception à 3600 enfants de pères prenant de la lamotrigine ou du lévétiracétam. Cette étude n’a pas montré de risque augmenté d’anomalies congénitales chez les enfants de pères exposés au valproate. Par contre, une augmentation du risque de troubles neurodéveloppementaux a été rapportée avec environ 6% d’enfants (de pères exposés) atteints contre 3% des enfants de pères traités par lamotrigine ou lévétiracétam (HR 1,5 ; IC95% : 1,1, 2,1) (1). Cependant, l’EMA, ayant noté des erreurs et de nombreuses limites dans cette étude, a demandé aux auteurs des informations supplémentaires et des modifications dans la méthode. En attente de ces nouveaux résultats, l’EMA recommande des mesures de précaution. En France, en 2023, l’ANSM a relayé cette information aux professionnels de santé, en appelant à la prudence et en demandant que soit remis aux patients un feuillet d’information lors de toute prescription ou délivrance de médicaments à base de valproate.
Depuis cette alerte, que s’est-il passé ? Existe-t-il de nouvelles données ?
A ce jour, nous n’avons aucune information sur les corrections et nouveaux résultats de l’étude commanditée par l’EMA. Néanmoins, deux publications (émanant aussi d’équipes scandinaves et utilisant les mêmes sources de données) apportent des éléments de réflexion complémentaires. La première, suédoise, publiée en 2020 (avant l’alerte), comparant 2087 enfants conçus par des pères exposés à des antiépileptiques à des enfants de pères non exposés (2) n’a pas retrouvé d’association entre l’exposition paternelle aux antiépileptiques, et en particulier au valproate (n=458), au moment de la conception et la survenue de troubles du neurodéveloppement (troubles autistiques, troubles déficitaires de l’attention, déficit intellectuel) chez leurs enfants. La deuxième étude (3), danoise, a été publiée en 2024, après l’alerte de l’EMA et de l’ANSM. Les auteurs ont comparé 1336 enfants de pères exposés au valproate au moment de la conception à plus d’un million d’enfants de pères non exposés et à 1663 enfants de pères exposés à la lamotrigine. L’analyse des données n’a pas mis en évidence d’augmentation du risque de troubles du neurodéveloppement chez les enfants de pères exposés au valproate. Pour essayer de pallier les biais évoqués dans l’étude ayant conduit à l’alerte (biais d’indication, absence de certaines données telles que génétiques, …), les auteurs de cette dernière étude ont utilisé différentes méthodes d’analyse (étude de siblings, modèle de Cox, effet dose…). Elles ont toutes conduit à des résultats similaires ne mettant pas en évidence de lien entre l’exposition paternelle au valproate et les troubles du neurodéveloppement dans la descendance.
Ces deux dernières études comportent également des limites et incluent des effectifs qui restent encore limités. Néanmoins, elles ne confirment pas le risque de troubles du neurodéveloppement chez les enfants de pères traités par valproate. En pratique, les données, publiées à ce jour, insuffisantes mais non inquiétantes, ne justifient pas la nécessité d’arrêter ou de changer un traitement par valproate chez un homme souhaitant concevoir.
1. catalogues.ema.europa.eu/system/files/2024-02/Valproate_PASS_Abstract_V2.0_0.pdf
2. Tomson T, Muraca G, Razaz N. Paternal exposure to antiepileptic drugs and offspring outcomes: a nationwide population-based cohort study in Sweden. J Neurol Neurosurg Psychiatry. 2020; 91(9):907-913.
3. Christensen J, Trabjerg BB, Dreier JW. Valproate Use During Spermatogenesis and Risk to Offspring. JAMA Netw Open. 2024; 7(6):e2414709.
Anti-inflammatoires non steroïdiens et grossesse : ne pas baisser la garde
Marie-Andrée Thompson-Bos, Cosette Le Souder (Montpellier), Philippe Deruelle (Gynécologie-Obstétrique, CHU Montpellier)
Deux patientes enceintes, respectivement à 32 et 34 SA, ont été récemment prises en charge par le service de gynécologie obstétrique de notre CHU pour surveillance, après prise d’AINS sur prescription médicale : kétoprofène, 100 mg, 2 prises pour otite externe dans le 1er cas et kétoprofène, 50 mg, 2 prises pour abcès dentaire dans le second. Un suivi échographique a été mis en place : liquide amniotique, dopplers fœtaux, échocardiographie avec contrôle à J15. L’évolution de ces deux grossesses est pour l’heure favorable.
Rappelons que les AINS sont formellement contre-indiqués à partir de 24 SA : toxicité rénale fœtale et cardio-pulmonaire (notamment constriction du canal artériel in utero, risque augmentant près du terme). La possibilité d’atteinte rénale existe dès 12 SA, début de la diurèse fœtale, particulièrement lors d’exposition prolongée. Au 1er trimestre, les AINS sont suspectés de pouvoir interférer avec l’implantation, par leur action inhibitrice de la synthèse des prostaglandines et pour certains, d’augmenter le risque de malformations cardiaques et de la paroi abdominale.
Une analyse détaillée avait été réalisée début 2023 par l’ANSM à partir des données de la Base Nationale de Pharmacovigilance et des résultats d’une étude issue du registre mère-enfants d’EPI-PHARE (à partir du Système National des Données de Santé) : entre 2018 et 2021, 11,7% des grossesses étaient exposées à un AINS (au moins une délivrance), dont 0,6% au-delà du 6ème mois. L’ANSM a procédé par la suite à une révision des RCP, à la fois au niveau de la rubrique « Mise en garde et précautions d’emploi » et « Fertilité/grossesse/allaitement » pour insister sur les risques et les préciser. La notice patient a également été mise à jour.
Pour faire simple, les AINS sont à bannir au cours de la grossesse. Selon l’indication, les corticoïdes (prednisone, prednisolone en priorité) peuvent être utilisés quel que soit le trimestre, avec une surveillance de principe de la mère et du nouveau-né en cas de traitement prolongé, de même que pour le paracétamol et les opiacés, avec quelques précautions (posologie et durée minimale efficace, surveillance du nouveau-né si utilisation près du terme)

BIP Occitanie fait partie de l'International Society of Drug Bulletins (ISDB)