

BIP 2025 N°2

EDITORIAL
Une rupture d’approvisionnement d’un médicament est définie comme une incapacité pour une pharmacie d’officine ou une pharmacie à usage intérieur d’un établissement (de santé ou médico-social) à dispenser un médicament à un patient dans un délai de 72 heures. Selon la DREES (27/3/25), en 2021 et 2022, le nombre de spécialités en rupture de stock a fortement augmenté, dépassant largement les précédents épisodes, notamment celui intervenu lors du premier confinement. Un pic de criticité a été atteint à l’hiver 2022-2023, avec environ 800 présentations médicamenteuses en rupture de stock simultanément. Le nombre de présentations en rupture a ensuite reflué progressivement. Au 31 décembre 2024, on comptait encore 400 présentations en rupture de stock. Depuis le 1er janvier 2025, 14 tensions d’approvisionnement et ruptures de stock en médicaments psychiatriques ont été publiées (lithium, quétiapine, sertraline, venlafaxine, olanzapine,…), manque de médicaments pour une discipline en manque de moyens, générant un déséquilibre accru pour les patients. Cette situation anxiogène est un véritable « casse-tête » pour les patients et les professionnels de santé. Par ailleurs, l’étude sur les conséquences iatrogènes des ruptures de stock (CIRUPT) menées par les CRPV entre 2004 et 2019 avait noté un phénomène en croissance avec 462 cas d’effets indésirables dont la moitié était grave. Fin 2024, onze fabricants de médicaments ont été sanctionnés par l’Agence du médicament (ANSM) pour ne pas avoir constitué de stocks suffisants de médicaments d’intérêt thérapeutique majeur (MITM), la majorité concernant les laboratoires fabriquant des génériques. Certes l’intensification des sanctions peut inciter les laboratoires à rester plus vigilants mais suffira-t-elle à calmer le phénomène des pénuries ? Pas sûr… Concentration des lignes de production en Chine ainsi qu’en Inde et augmentation de la demande mondiale en médicaments et vaccins ne jouent pas en faveur d’une amélioration rapide. Néanmoins, on peut s’interroger sur l’intérêt des laboratoires détenteurs des AMM de médicaments onéreux, à régler la sanction mais de privilégier leurs chaînes de fabrication pour les spécialités onéreuses (les anticorps monoclonaux indiqués dans les pathologies cancéreuses ou maladies immunologiques). A l’instar de la liste des médicaments essentiels de l’OMS, ne faut-il pas établir au niveau européen une liste rouge de principes actifs avec tolérance zéro de rupture de stock par les laboratoires fabricants ? Un vœu pieux !
Haleh Bagheri
Sommaire
- INFOS IMPORTANTES - ALERTES
- PHARMACOLOGIE CLINIQUE
- PHARMACOVIGILANCE
- PHARMACO-EPIDEMIOLOGIE
- MEDICAMENTS, REPRODUCTION ET PERINATALITE
- PHARMACOLOGIE SOCIALE
- ADDICTOVIGILANCE
Fezolinetant : nouveau traitement non hormonal des symptômes de la ménopause
Agathe Soto (étudiante), Jean-Luc Faillie (Montpellier)
Avant la ménopause, le centre thermorégulateur hypothalamique est finement réglé par l’équilibre entre le système œstrogénique et la neurokinine B (NKB) au sein des neurones KNDy (kisspeptine/NKB/dynorphine). En périménopause, la chute des œstrogènes entraîne une hyperactivité de ces neurones, à l’origine des bouffées de chaleur. Ce phénomène peut être atténué par un antagoniste du récepteur NK3 de la NKB, le fezolinetant.
Commercialisé en France depuis le 8 avril 2025, le fezolinetant (VEOZA®) est un traitement non hormonal indiqué pour les symptômes vasomoteurs modérés à sévères de la ménopause (1). Sa place dans l’arsenal thérapeutique reste à préciser, mais il pourrait bénéficier aux patientes chez qui un THS œstrogénique est contre-indiqué (antécédent de cancer du sein, risque thromboembolique).
Les effets indésirables les plus fréquents sont diarrhée, insomnie et élévation des transaminases. En post-commercialisation aux États-Unis, des cas d’atteintes hépatiques graves ont été rapportés. Après analyse par l’EMA, une communication de l’ANSM, publiée moins d’un mois après sa mise sur le marché en France, recommande de renforcer la surveillance hépatique : un bilan hépatique doit être réalisé avant tout démarrage, puis mensuellement pendant les trois premiers mois de traitement (2). Le fezolinetant doit être interrompu immédiatement en cas de signes cliniques ou biologiques d’atteinte hépatique. Il convient d’informer la patiente des symptômes évocateurs : fatigue, prurit, ictère, urines foncées, selles décolorées, nausées, vomissements, anorexie et/ou douleurs abdominales.
Comme pour tout nouveau médicament, il est essentiel de vérifier que l’efficacité observée en essai se confirme cliniquement en vie réelle, de surveiller attentivement le profil de sécurité notamment à long terme et de signaler au CRPV tout effet indésirable suspect.
- https://www.ema.europa.eu/fr/documents/product-information/veoza-epar-product-information_fr.pdf
- https://ansm.sante.fr/informations-de-securite/veoza-fezolinetant-risque-de-lesion-hepatique-et-nouvelles-recommandations-concernant-la-surveillance-de-la-fonction-hepatique-avant-et-pendant-le-traitement
TRAITEMENT DE LA TEIGNE CHEZ L’ENFANT DEPUIS L’ARRÊT DE COMMERCIALISATION DE LA GRISEOFULVINE
Marilou Duboëlle (docteur junior en pharmacie) et Pascale Palassin (Montpellier)
La teigne est une dermatophytie fréquente qui affecte essentiellement les enfants et dont les dermatophytes en cause en France appartiennent aux genres Trichophyton et Microsporum. Pendant longtemps, la griséofulvine a été le seul traitement bénéficiant d'une Autorisation de Mise sur le Marché (AMM) pour les enfants, bien que sa balance bénéfices/risques ait été considérée comme suboptimale. Sa commercialisation a été interrompue en 2021 en France, laissant les prescripteurs sans solution thérapeutique chez les jeunes patients, puisque la terbinafine, alternative thérapeutique principale, est officiellement contre-indiquée chez l’enfant selon son Résumé des Caractéristiques du Produit (RCP). Cette situation soulève des interrogations quant aux options thérapeutiques pour une prise en charge efficace et sécuritaire de la teigne en pédiatrie.
Plusieurs sociétés savantes, coordonnées par le Centre de Preuves en Dermatologie (CDP), ont élaboré en 2021 un algorithme (1), mis à jour en décembre 2024, et des outils pratiques pour guider les prescripteurs, publiés dans les Annales de Dermatologie et disponibles sur le site du CDP. L'Agence Nationale de Sécurité du Médicament et des produits de santé (ANSM) a validé et intégré cet algorithme dans un communiqué du 9 juillet 2021 (2). Malgré sa contre-indication officielle chez les enfants, la terbinafine occupe ainsi une place centrale dans ces nouvelles recommandations.
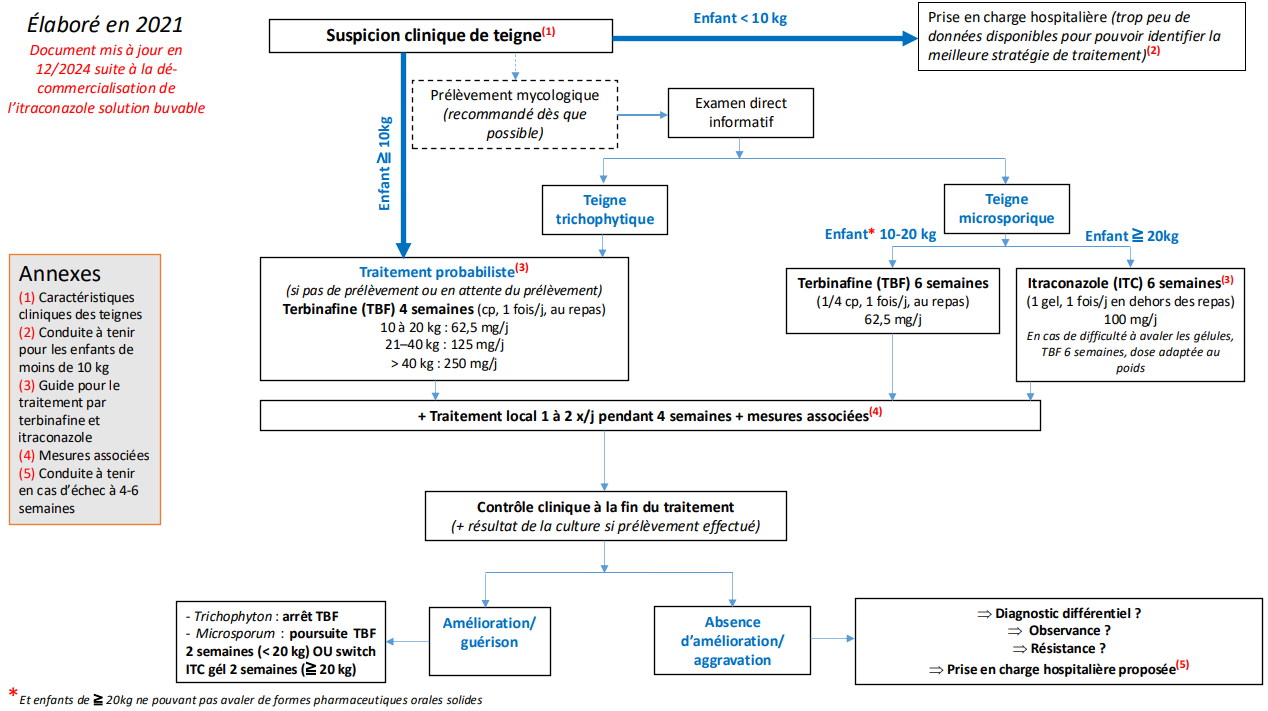
En parallèle, ces travaux ont conduit à la mise en place d'un Cadre de Prescription Compassionnelle (CPC) pour l'itraconazole, permettant aux médecins de prescrire cet antifongique aux enfants atteints de teigne à Microsporum, sous certaines conditions.
L’itraconazole, notamment sous forme de SPORANOX 10 mg/mL solution buvable et 100 mg gélule, est désormais disponible pour les enfants pesant au moins 10 kg, offrant ainsi une alternative précieuse. Ce CPC, valable trois ans, est accessible sur le site de l’ANSM et ne nécessite plus de prescription initiale hospitalière.
Quel que soit le traitement médicamenteux, les mesures d’hygiène restent indispensables pour éviter la contagion et une ré-infestation (désinfection de tout objet en contact avec l’enfant par nettoyage à haute température ou isolement pendant au moins 48 heures). Une prise en charge rapide et adaptée permet de prévenir le risque de complications, telles que cicatrices ou alopécies définitives, ou encore de récidive, dont l’absence doit être confirmée par un dermatologue ou un pédiatre.
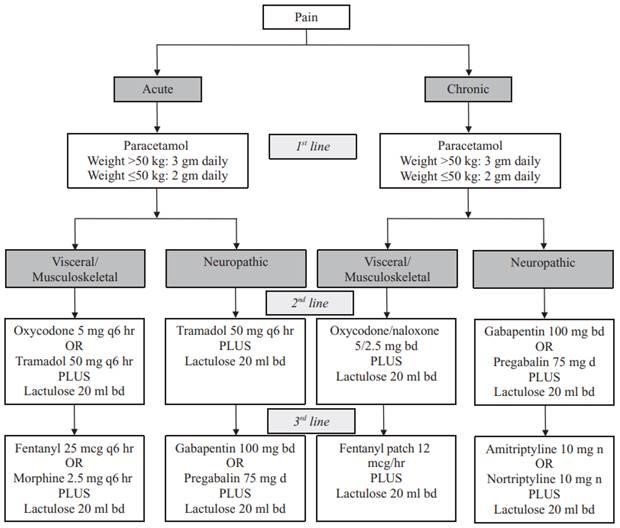
Le défi de la prise en charge de la douleur chronique chez le patient cirrhotique
Ariane Olivan et Pascale Palassin (Montpellier)
La prise en charge de la douleur chronique chez le patient cirrhotique est complexe en raison notamment des altérations pharmacocinétiques et pharmacodynamiques des médicaments induites par l’insuffisance hépatique, du risque d’encéphalopathie hépatique et du risque potentiel de pharmacodépendance.
Le médicament antalgique de première intention reste le paracétamol, en raison de son efficacité et de sa relative sécurité d’emploi. Il peut être utilisé chez le patient cirrhotique, à condition de limiter la dose à 2 g/jour chez les patients à risque de consommation d’alcool ou de malnutrition. (1,2,3)
Les anti-inflammatoires non stéroïdiens (AINS) restent fortement déconseillés, en raison de leur néphrotoxicité et du risque majoré d’hémorragies digestives qu’ils induisent en altérant la muqueuse gastrique par la réduction de synthèse des prostaglandines. (3)
Le tramadol n’apparaît pas comme une alternative de choix chez un patient sujet à la dépendance avec un risque important d’addiction. L’usage des dérivés opiacés est délicat en raison du risque d’abus et d’augmentation du risque d’encéphalopathie hépatique. Toutefois, certaines études suggèrent une tolérance comparable à celle des patients non cirrhotiques, sous réserve d’une adaptation posologique (4,5). La morphine, dont le métabolisme est altéré en cas d’insuffisance hépatique sévère, doit être administrée à doses réduites et espacées. En revanche, la méthadone pourrait constituer une alternative plus sûre en cas de douleur chronique et de dépendance aux opiacés.
La gabapentine et la prégabaline peuvent être envisagées comme des alternatives de choix en cas de douleurs neuropathiques, en raison de l’absence de métabolisme hépatique. (3)
Dans tous les cas, la prescription d’un antalgique à visée chronique nécessite une évaluation de la douleur permettant d’orienter le choix thérapeutique. Quelle que soit l’option choisie, il convient d’effectuer une titration lente, en prescrivant bien souvent des doses plus faibles et espacées que chez le patient non cirrhotique. (3) Cet article de revue propose un arbre décisionnel pour la prise en charge de la douleur chronique chez les patients atteints de pathologie hépatique chronique :
- Chandok N, Watt KD. Pain management in the cirrhotic patient: the clinical challenge. Mayo Clin Proc. 2010 May;85(5):451-8.
- Bosilkovska M, Walder B, Besson M, Daali Y, Desmeules J. Analgesics in patients with hepatic impairment: pharmacology and clinical implications. Drugs. 2012 Aug 20;72(12):1645-69.
- Dwyer JP, Jayasekera C, Nicoll A. Analgesia for the cirrhotic patient: a literature review and recommendations. J Gastroenterol Hepatol. 2014;29(7):1356-60.
- Tovoli F, De Lorenzo S, Samolsky Dekel BG, Piscaglia F, Benevento F, Brandi G, Bolondi L. Oral oxycodone/naloxone for pain control in cirrhosis: Observational study in patients with symptomatic metastatic hepatocellular carcinoma. Liver Int. 2018 Feb;38(2):278-284.
- Ali B, Jiang Y, Agbim U, Kedia SK, Satapathy SK, Barnes M, Maliakkal B, Nair SP, Eason JD, Gonzalez HC. Effect of opioid treatment on clinical outcomes among cirrhotic patients in the United States. Clin Transplant. 2020 Jun;34(6):e13845.
Lécanémab : de nouvelles données de sécurité
Romain Barus (Toulouse)
Le lécanémab (LEQEMBI®) a finalement reçu une Autorisation de Mise sur le Marché (AMM) le 15 avril 2025 par l’EMA (European Medical Agency) dans la prise en charge des patients atteints de troubles cognitifs légers et de démence au stade léger (maladie d’Alzheimer débutante), non porteurs ou hétérozygotes pour l’allèle ε4 du gène de l’apolipoprotéine E (ApoE ε4) et présentant une pathologie amyloïde confirmée. Il s’agit d’un anticorps dirigé contre les formes agrégées solubles et insolubles du peptide béta-amyloide qui diminue la formation de plaques amyloïdes.
Cette AMM se base notamment sur l’évaluation post-hoc par l’EMA des données de l’essai clinique CLARITY-AD (1) : après exclusion des patients homozygotes du gène ApoE ε4 en raison d’un risque important d'anomalies d'imagerie liées à l'amyloïde (ARIA) favorisant un œdème cérébral (ARIA-E) ou des hémorragies cérébrales (ARIA-H), l’augmentation du score moyen CDR-SB évaluant les fonctions cognitives (Clinical dementia Rating-Sum of Boxes, variant de 0 - pas d’atteinte à 18 - atteinte maximale) était de 1,22 dans le groupe lécanémab après 18 mois versus 1,76 dans le groupe placebo soit une différence de 0,54.
Aux Etats-Unis, l’indication du lécanémab ne tient pas compte du statut ApoE ε4. Un centre mémoire de Washington a récemment publié son retour d’expérience sur la faisabilité et la sécurité de ce médicament (2). Sur la période d’août 2023 à octobre 2024, 3938 patients ont été reçu dans ce centre mémoire. Deux centre trente-quatre d’entre eux (5,9%) ont reçu du lécanémab dont la moitié étaient des femmes, 8,5% des patients étaient homozygotes pour le gène ApoE ε4. La durée moyenne de suivi de traitement était de 6,5 (+/- 3,2) mois. Trente-sept pour cent des patients ont présenté des réactions à la perfusion (frissons, céphalées, nausées et fièvre), surtout lors des deux premières injections. Les patients ayant reçu moins de 4 perfusions n’ont pas présenté d’ARIA. Sur les 194 patients ayant reçu au moins 4 perfusions, 42 (22%) ont présenté des ARIA (29 des ARIA-E et 13 des ARIA-H). Les ARIA-E survenaient dans les premiers mois de traitement et rarement après 6 mois de traitement. Au contraire, la prévalence des ARIA-H augmentait au cours du temps. A 12 mois de traitement, 29% (17% ARIA-E et 13% d’ARIA-H) des patients présentaient des ARIA. Les ARIA-E étaient plus fréquentes chez les sujets homozygotes ApoE ε4 que chez les sujets hétérozygotes ApoE ε4 et non porteur du gène ApoE ε4. Les ARIA-H étaient plus fréquentes chez les sujets avec un nombre élevé de microhémorragies cérébrales. Aucun décès ou macrohémorragie n’a été observée. Enfin, l’augmentation du score CDR-SB au cours de la période de traitement était de 1,11 par an, correspondant à l’évolution de ce score dans le groupe placebo de l'étude CLARITY-AD.
Tandis que ce médicament n’a pas encore été évalué par la commission de transparence de la Haute autorité de santé, les recommandations de la fédération française des centres mémoires concernant son utilisation sont déjà disponibles (3). Les auteurs sont en faveur de ne pas initier le lécanémab chez les patients ayant une angiopathie amyloïde cérébrale probable (ces patients présentent un risque hémorragique élevé) et également de réaliser une IRM avant la 3ème perfusion (soit plus précocement qu’indiqué dans la monographie du médicament en raison du risque d’ARIA survenant avant la 5ème perfusion). Enfin, il est également discuté d’un protocole d’arrêt de ce médicament après 18 mois de traitement.
1. Dyck CH van, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, et al. Lecanemab in Early Alzheimer’s Disease. N Engl J Med. 4 janv 2023;388(1):9‑21.
2. Paczynski M, Hofmann A, Posey Z, Gregersen M, Rudman M, Ellington D, et al. Lecanemab Treatment in a Specialty Memory Clinic. JAMA Neurol. 12 mai 2025; doi.org/10.1001/jamaneurol.2025.1232
3. Villain N, Planche V, Lilamand M, Cordonnier C, Soto-Martin M, Mollion H, et al. Lecanemab for early Alzheimer’s disease: Appropriate use recommendations from the French federation of memory clinics. J Prev Alzheimers Dis. 1 avr 2025;12(4):100094.
Amphotéricine B injectable : confusion entre formes pharmaceutiques, un risque d’erreur médicamenteuse potentiellement fatale
Pauline Schiro (Toulouse)
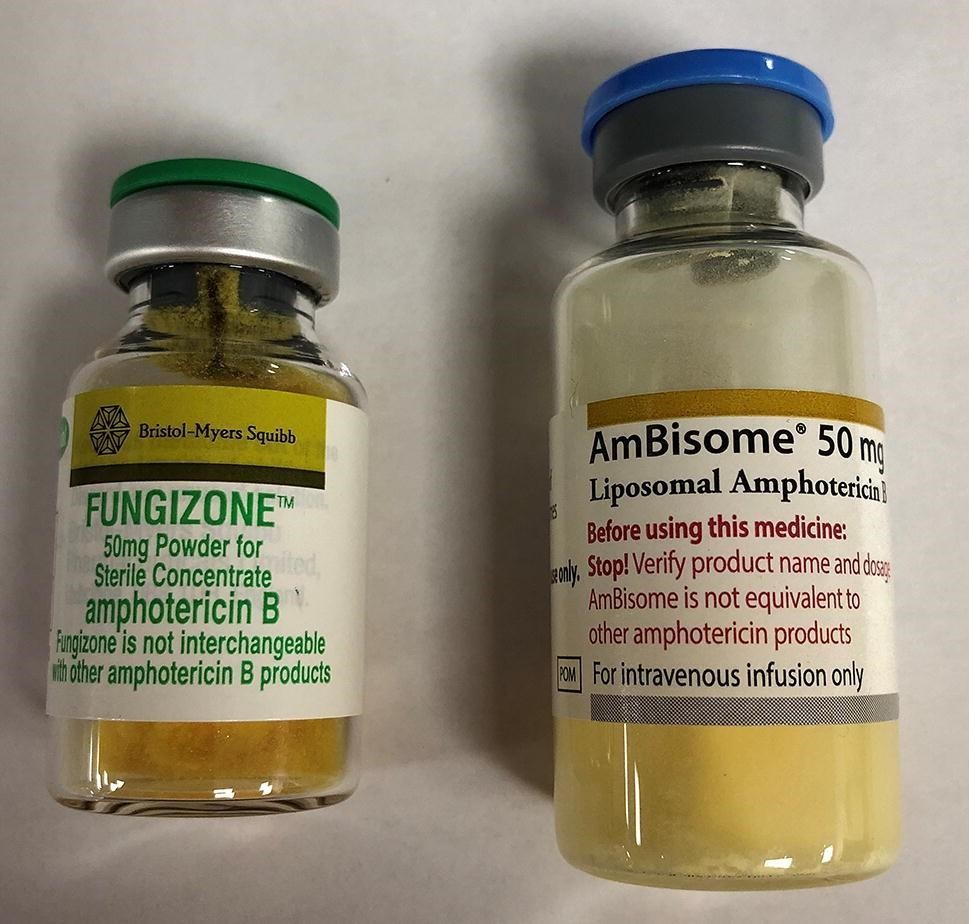
L'amphotéricine B est un antibiotique antifongique de la famille des macrolides polyènes, disponible en France sous deux formes injectables : liposomale (AmBisome®) et conventionnelle (Fungizone®). Bien que partageant le même principe actif et spectre antifongique, ces formulations ne sont pas interchangeables. Elles diffèrent par leur mode de préparation, d’administration et surtout par leur posologie : 3 mg/kg/jour pour AmBisome® vs 0,3 à 1,5 mg/kg/jour pour Fungizone®. Par ailleurs, malgré des posologies distinctes, toutes deux se présentent en fioles de 50 mg.
Cette similarité expose à un risque d’erreur médicamenteuse. L'une des plus fréquentes est l'administration de Fungizone®, au dosage et au débit d'AmBisome®,entrainant un surdosage en amphotéricine B, pouvant causer de graves atteintes cardiaques ou rénales.
En 2017, l’ANSM a alerté les professionnels de santé sur ce risque via une lettre d’information, recommandant de prescrire les médicaments sous leurs noms de marque en plus de la dénomination commune internationale (DCI) et de vérifier les paramétrageslocaux des logiciels d’aide à la prescription (1).
Ce risque est toujours d’actualité. Un cas récent l’illustre : un patient hospitalisé s’est vu prescrire de l’amphotéricine B liposomale (AmBisome®), mais une confusion a conduit à l’injection de la forme conventionnelle (Fungizone®). Le patient a reçu une dose dix fois supérieuresur une durée de perfusion plus courte (1 heure au lieu de 2). Ce surdosage a entrainé des troubles du rythme cardiaque qui ont conduit à son décès. Une dizaine de cas de confusion ont été déclarées postérieure à l’alerte de l’ANSM.
En conclusion, il est nécessaire de sensibiliser les équipes médicales et de renforcer la vigilance à toutes les étapes : prescription, dispensation, préparation et administration.
La mode du CBD : réalités pharmacologiques
Laure Henry (externe en pharmacie), Christelle Philibert (Montpellier)

Le cannabidiol (CBD) est un phytocannabinoïde issu du chanvre tout comme le tétrahydrocannabinol (THC). On le trouve principalement dans les fleurs et les feuilles. L’arrêté du 30 décembre 2021 prévoit que la teneur en THC des produits obtenus à partir du chanvre ne soit pas supérieure à 0,3%, tous les produits contenant plus de 0,3 % de THC sont considérés comme des stupéfiants et donc, à ce titre, interdits.
Si le CBD n’agit pas ou très peu sur les récepteurs cannabinoïdes (ceux où se fixe le THC), son action sur les récepteurs dopaminergiques et sérotoninergiques en fait un produit psychoactif à part entière.
En France, une expérimentation sur l’usage thérapeutique du cannabis est en cours depuis 2021. Cette expérimentation porte sur un nombre restreint d’indications : douleurs neuropathiques réfractaires aux thérapies accessibles (médicamenteuses ou non) ; certaines formes d’épilepsie sévères et pharmaco-résistantes ; certains symptômes rebelles en oncologie liés au cancer ou à ses traitements ; situations palliatives ; spasticité douloureuse de la sclérose en plaques ou des autres pathologies du système nerveux central. Il n’existe sur le marché qu’un seul médicament à base de CBD ayant obtenu une AMM pour traiter les crises convulsives de syndromes épileptiques rares: Epidyolex®.
En dehors de cette expérimentation, le CBD est légalement incorporé dans différents produits : e-liquides pour cigarettes électroniques, compléments alimentaires, produits diététiques ou de régimes, produits cosmétiques. Les produits contenant du CBD ne peuvent être commercialisés sans évaluation préalable et autorisation par l’Autorité européenne de sécurité des aliments (EFSA).
On l’aura compris, la consommation de ces produits n’est toutefois pas anodine, premièrement du fait de l’action dépresseur du système nerveux central, de son effet hépatotoxique (notamment en association à des médicaments hépatotoxiques ou en cas d’insuffisance hépatique) et des troubles digestifs qu’il peut entraîner.
Cet effet dépresseur du SNC peut être à l’origine d’interactions pharmacodynamiques avec d’autres traitements dépresseurs comme les antalgiques morphiniques, les antiépileptiques, les médicaments sédatifs, anxiolytiques/hypnotiques potentialisant la sédation, les risques de chutes et la somnolence.
Le CBD est de plus un inhibiteur puissant de plusieurs cytochromes enzymatiques et notamment des cytochromes CYP 2B6, 2C19 et 3A4, entraînant des interactions pharmacocinétiques avec de nombreux médicaments comme des antiépileptiques, des anticoagulants, des immunosuppresseurs, des opiacés ou encore des antidépresseurs.
Le CBD n’est pas un stupéfiant (contrairement au THC) mais reste une substance psychoactive qui peut avoir des interactions avec d’autres molécules notamment avec certains médicaments. Actuellement, toutes ces interactions et l’importance de leurs impacts cliniques ou biologiques ne sont pas encore bien connues. Il est donc nécessaire de rester très prudent sur l’association du CBD avec d’autres traitements chroniques en cours.
1. Association Française des centres d’Addictovigilance : Pr Joëlle MICALLEF, Dr Anne BATISSE, Dr Bruno REVOL,. Pharmacologie du Cannabidiol : Points de vigilance, conséquences et risques chez l’homme. 2021.
2.https://www.urps-pharmaciens-hdf.fr/wp-content/uploads/2024/02/2023-01-11-fiche-CBD-VF.pdf. Qu’est-ce que le CBD ? Où en trouve-t-on ? Est-il « légal» ? 2023.
3..https://www.ordre.pharmacien.fr/les-communications/focus-sur/les-actualites/point-sur-les-produits-contenant-du-cannabidiol-cbd. Point sur les produits contenant du cannabidiol (CBD) | CNOP
Source : ANSM [Internet]. [cité 22 mai 2025]. Actualité - Mélanger CBD et médicaments, ce n’est jamais anodin. Disponible sur : https://ansm.sante.fr/actualites/melanger-cbd-et-medicaments-ce-nest-jamais-anodin

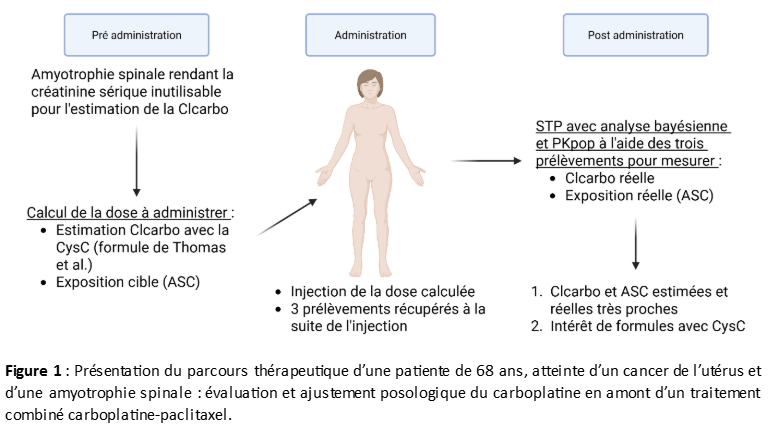
Suivi Thérapeutique Pharmacologique du carboplatine : adaptation de dose basée sur la cystatine C seule.
Matthieu Gracia, Laurence Gladieff, Etienne Chatelut, Fabienne Thomas (Toulouse)
Le carboplatine est une molécule de chimiothérapie utilisée dans de nombreuses situations tumorales (cancer de l’ovaire, de l’utérus, des poumons, de la tête et du cou …). L’élimination, principalement rénale par filtration glomérulaire, ainsi que la forte variabilité interindividuelle, sont deux paramètres qui expliquent l’intérêt du suivi thérapeutique pharmacologique (STP) pour cette molécule dans certaines situations (protocoles haute dose, patients avec caractéristiques morphologiques extrêmes).
Une des particularités de prescription de cette molécule est étroitement liée au fait qu’il est possible d’estimer la clairance du carboplatine à partir du DFG ou de formules dédiées [1–4]. La dose recommandée de carboplatine est donc calculée à partir de la clairance estimée du carboplatine (Clcarbo) et d’une exposition cible qui suit les recommandations (ASC cible). Habituellement, l’estimation de la Clcarbo s’effectue selon la formule de Calvert comme suit : Clcarbo (ml/min) = Débit de Filtration Glomérulaire + 25 [1]. Le débit de filtration glomérulaire (DFG) peut être calculé par des formules, comme celle de Calvert CKD-EPI, qui prennent en considération la créatinine sérique (Scr) et des paramètres propres au patient comme l’âge, le sexe et la surface corporelle [2]. Plus récemment, de nouvelles formules sont apparues en se basant sur la Scr couplée à la cystatine C (CysC) [3] ou des formules basées sur la CysC seule [4].
Nous décrivons ici le cas particulier d’une patiente âgée de 68 ans atteinte d’un cancer de l’utérus et d’une amyotrophie spinale pour laquelle un calcul de dose de carboplatine avant son traitement par association carboplatine-paclitaxel a dû être effectué. Son amyotrophie spinale a rendu ininterprétable sa valeur de créatinine sérique (mesurée à 10 µmol/L) car non représentative de sa fonction rénale (DFG largement surestimé). Nous avons donc utilisé la formule de Thomas et al., basée sur la CysC de cette patiente ainsi que le poids (BW) et l’âge, afin d’estimer son DFG [4]. Cette formule nous a permis d’en déduire la Clcarbo (Clcarbo = 99.(CysC/1.0)-0.573.(BW/65)0.460.(age/56)-0.405) et, couplée à l’ASC cible, nous avons calculé la dose de carboplatine à administrer.
Selon notre recommandation de dose, la patiente a bien toléré le cycle de chimiothérapie. De plus, le suivi thérapeutique pharmacologique (STP), basé sur 3 prélèvements sanguins post-administration et une approche Bayésienne, nous a permis de mesurer la Clcarbo et l’ASC réelles. La Clcarbo et l’ASC mesurées avec le STP se sont avérées très proches respectivement de la Clcarbo estimée par la formule de Thomas et al. et de l’ASC cible, toutes deux définies avant l’administration.
Ainsi, ce cas montre l’importance de disposer de formules intégrant la CysC seule afin d’être en capacité de répondre à toutes les demandes et cela même pour des patients avec des caractéristiques physiologiques inhabituelles. Enfin, l’intérêt du STP est, une nouvelle fois, renforcé pour optimiser l’efficacité et la tolérance thérapeutique du carboplatine.
1. Calvert AH, Newell DR, Gumbrell LA, et al (1989) Carboplatin dosage: prospective evaluation of a simple formula based on renal function. J Clin Oncol 7:1748–1756. doi.org/10.1200/JCO.1989.7.11.1748
2. Levey AS, Stevens LA, Schmid CH, et al (2009) A New Equation to Estimate Glomerular Filtration Rate. Annals of Internal Medicine 150:604–612. doi.org/10.7326/0003-4819-150-9-200905050-00006
3. Inker LA, Schmid CH, Tighiouart H, et al (2012) Estimating Glomerular Filtration Rate from Serum Creatinine and Cystatin C. N Engl J Med 367:20–29. doi.org/10.1056/NEJMoa1114248
4. Thomas F, Séronie-Vivien S, Gladieff L, et al (2005) Cystatin C as a New Covariate to Predict Renal Elimination of Drugs: Application to Carboplatin. Clinical Pharmacokinetics 44:1305–1316. doi.org/10.2165/00003088-200544120-00009

BIP Occitanie fait partie de l'International Society of Drug Bulletins (ISDB)