
BIP 2026 N°2

Editorial
1976-2026 : la pharmacovigilance a 50 ans !
Le Réseau Français des Centres Régionaux de Pharmacovigilance (RFCRPV) a fêté ses 50 ans le 2 juin dernier. Cela a été l’occasion de réunir les membres actuels des CRPV et le plaisir de retrouver les plus anciens qui ont tant apporté à notre réseau !
Le Réseau, au fil des décennies, s’est structuré et renforcé formant aujourd’hui un maillage territorial solide et performant pour capter les signaux de sécurité (y compris faibles), avec une expertise unique.
Notre travail dépasse largement le simple recensement d’effets indésirables et la rédaction d’expertises. Au sein de chacune de nos régions et de nos établissements, nous participons activement à la prévention, à l’information et la formation sur la iatrogénèse médicamenteuse et le bon usage des médicaments. Au fil de ces 50 années, plusieurs éléments ont ébranlé ou marqué l’histoire de la pharmacovigilance, de nos centres et de notre réseau (Médiator®, Lévothyrox®, COVID-19). Mais ce réseau a su démontrer sa robustesse, son adaptabilité et sa résilience.
Cette journée a été l’occasion de redécouvrir l’histoire de la pharmacovigilance française depuis les années 70, organisation qui a été un modèle pour les autres pays. Plusieurs problématiques actuelles ont été abordées, notamment l’importance du rôle du RFCRPV dans la lutte contre la désinformation et sa place légitime dans le bon usage du médicament.
Une table ronde, réunissant Mme la Directrice Générale de l’ANSM, des pharmacologues, un représentant d’association de patients et la Revue Prescrire, a permis d’échanger et de réfléchir au sujet très actuel de « l’efficience dans la surveillance des médicaments ». Les échanges ont été très riches et ont permis de faire le point sur les attentes actuelles en termes de prévention et prise en charge de la iatrogénie médicamenteuse, domaine de compétence des CRPV. Il a été souligné l’importance de renforcer l’existant plutôt que de se disperser dans de nouvelles structures ou organisations, ce qui serait contreproductif et source de dépenses inutiles. Cela risquerait de brouiller le message et de faire perdre confiance dans un système qui a pourtant fait ses preuves !
La pharmacovigilance française forte de ses 50 ans d’expertise est prête pour affronter l’avenir et les nouveaux défis, en restant toujours au service des professionnels de santé et des patients.
Sommaire
- INFORMATIONS IMPORTANTES - ALERTES
- PHARMACOLOGIE CLINIQUE
- PHARMACOVIGILANCE
- MEDICAMENTS, REPRODUCTION ET PERINATALITE
- PHARMACOLOGIE SOCIALE
- ECOPHARMACOLOGIE
- ADDICTOVIGILANCE
- PHARMACOLOGIE-TOXICOLOGIE
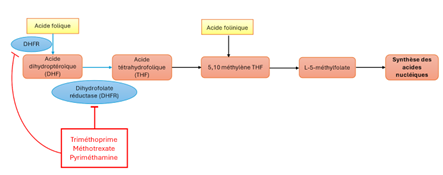
Supplémentation en acide folique / acide folinique lors des traitements par inhibiteurs de la DHFR : quelle place en pratique ?
Joseph Lenoir (interne), Marilou Duboëlle (AHU) (Montpellier)
Plusieurs médicaments exercent leur effet en inhibant la dihydrofolate réductase (DHFR), enzyme clé du métabolisme des folates impliquée dans la synthèse des acides nucléiques (cf. schéma). C’est notamment le cas du méthotrexate, du triméthoprime (composant du cotrimoxazole) ou encore de la pyriméthamine. L’inhibition de cette enzyme peut entraîner une carence fonctionnelle en folates, susceptible d’être à responsable de toxicités, en particulier hématologiques. Dans ce contexte, la place d’une supplémentation en acide folique ou en acide folinique est régulièrement discutée.
L’acide folique doit être converti par la DHFR pour devenir biologiquement actif, ce qui limite son efficacité en cas d’inhibition de cette enzyme. À l’inverse, l’acide folinique correspond à une forme réduite et active des folates, directement utilisable par les voies métaboliques, permettant de contourner partiellement le blocage enzymatique induits par les inhibiteurs de la DHFR. Sur le plan pharmacologique, il apparaît donc plus adapté pour corriger une toxicité induite par ces médicaments, son activité étant indépendante de cette étape enzymatique.
Les indications officielles de l’acide folinique incluent la prévention et le traitement des toxicités hématologiques induites par les médicaments inhibant la DHFR. L’acide folique, quant à lui, est principalement indiqué dans la prévention des anomalies de fermeture du tube neural en période périconceptionnelle, ainsi que dans la prise en charge des carences en folates responsables d’anémies macrocytaires ou liées à des troubles de l’absorption intestinale, quelle qu’en soit l’origine.
Ainsi, les données actuellement disponibles suggèrent que l’acide folinique dispose d’une indication reconnue pour la correction d’une hématotoxicité induite par les médicaments inhibant la DHFR. En revanche, une supplémentation prophylactique systématique ne paraît pas justifiée. Dans le cadre d’une antibiothérapie par cotrimoxazole, une supplémentation préventive paraît plutôt devoir être réservée à certaines situations à risque, notamment en cas de traitement prolongé (> 15 jours), de cytopénie préexistante ou de terrain particulièrement exposé à une toxicité hématologique. À l’inverse, l’acide folique ne dispose pas actuellement de données robustes permettant de soutenir son utilisation dans cette indication spécifique, tout en conservant ses indications propres.
Néanmoins, certaines données invitent à nuancer la distinction entre acide folique et acide folinique en prophylaxie. Plusieurs travaux suggèrent en effet un risque théorique d’altération de l’efficacité anti-infectieuse de certains inhibiteurs de la DHFR lors de l’administration concomitante d’acide folinique, notamment avec le cotrimoxazole utilisé dans le traitement de la pneumocystose. À l’inverse, des données limitées issues d’utilisations prolongées d’autres inhibiteurs de la DHFR suggèrent une efficacité relativement comparable de l’acide folique et de l’acide folinique dans la prévention de la toxicité hématologique. Ces éléments demeurent toutefois fragiles, hétérogènes et difficilement extrapolables aux différentes situations cliniques, en particulier lorsque l’efficacité du traitement inhibiteur de la DHFR constitue un enjeu thérapeutique majeur.
Les analogues GLP-1 passent à l'oral
Paul MATHIEU (Interne), Jean-Luc FAILLIE (Montpellier)
L’obésité touche près de 2,5 milliards de personnes dans le monde et constitue un enjeu majeur de santé publique. Les agonistes des récepteurs au GLP-1 ont transformé sa prise en charge, mais leur administration injectable peut limiter l’adhésion au traitement. L’orforglipron est le premier analogue oral non peptidique du GLP-1, administré une fois par jour sans condition particulière de prise (horaire ou alimentation), développé comme alternative potentiellement plus accessible aux formulations injectables.
L’essai de phase 3 ATTAIN-11 a évalué l’efficacité et la sécurité de l’orforglipron chez des adultes obèses sans diabète de type 2. Dans cette étude randomisée contre placebo, les patients (IMC > 30 kg/m² ou ≥ 27 kg/m² avec comorbidité) recevaient 6 mg, 12 mg ou 36 mg/j d’orforglipron, en complément de mesures hygiéno-diététiques. Après 72 semaines, la perte de poids moyenne supplémentaire versus placebo variait de -5,5 % à -9 %, avec un effet dose-dépendant. Sous 36 mg/j, 54,6 % des patients ont perdu au moins 10 % de leur poids (contre 12,9 % sous placebo), plus d’un tiers au moins 15 % et près d’un cinquième au moins 20 %. Des améliorations significatives du tour de taille, de la pression artérielle et du profil lipidique ont également été observées, suggérant un bénéfice cardiométabolique potentiel.
Le profil de tolérabilité était conforme à celui de la classe des agonistes du GLP-1, principalement marqué par des effets indésirables gastro-intestinaux. Aucun signal de toxicité hépatique n’a été identifié, contrairement à certaines molécules de développement antérieur.
Ainsi, l’orforglipron est présentée comme une alternative orale prometteuse aux agonistes du GLP-1 injectables, avec une efficacité significative sur la perte de poids bien que légèrement inférieure à celle des formes injectables et sur les facteurs de risque cardiométabolique bien que son bénéfice clinique cardiovasculaire reste toutefois à démontrer.
Ces données ont conduit à l’approbation de FOUNDAYO® par la FDA le 1er avril 2026, en association à un régime hypocalorique et à l’activité physique chez les adultes obèses ou en surpoids avec comorbidité. La posologie approuvée2 repose sur une titration progressive de 0,8 mg à 17,2 mg/j à un prix allant de 149 à 399 dollars par mois3. En Europe, le développement clinique se poursuit avec l’essai ATTAIN-24 chez des patients pédiatriques atteints de diabète de type 2. À ce jour, aucune autorisation n’a été délivrée par l’EMA et le médicament n’est pas disponible en France. Toutefois, son éventuelle commercialisation à venir invite à anticiper les enjeux liés à son utilisation. Au-delà des bénéfices attendus pour les patients, sa formulation orale, plus accessible et plus facile d’emploi que les analogues injectables, pourrait favoriser une augmentation du mésusage avec les risques sanitaires qui en découlent.
- Sean Wharton, et al. Orforglipron, an Oral Small-Molecule GLP-1 Receptor Agonist for Obesity Treatment. N Engl J Med 2025;393:1796-806.
- FDA Approves First New Molecular Entity Under National Priority Voucher Program | FDA
- (Coverage & Savings: Self Pay & Savings Card | Foundayo™ (orforglipron), v02/06/2026)
- European Medicines Agency decision P/0178/2024 of 6 May 2024 on the agreement of a paediatric investigation plan and on the granting of a deferral and on the granting of a waiver for orforglipron, (EMEA-003299-PIP01-22)
N-acétylcystéine IV : un protocole complexe à sécuriser
Hanna Owsinski (interne), Marilou Duboëlle, Virginie Brès (Montpellier)
Le médicament HIDONAC (N-acétylcystéine, NAC) est utilisé comme antidote d’un surdosage en paracétamol. L’examen du RCP révèle un protocole particulièrement complexe, appelant à une vigilance renforcée face aux risques d’erreurs auxquels peuvent être confrontés les professionnels lors de son application.
Le protocole IV repose sur un schéma en trois perfusions successives, chacune caractérisée par des volumes, des doses et des durées d’administration différentes. Ce protocole est décrit plus précisément dans le tableau ci-dessous (données du RCP). La complexité de ce schéma nous est apparue constituer un facteur majeur de risque d’erreur, notamment en contexte d’urgence (1).
Schéma en trois perfusions délivrant au total 300mg/kg sur 21h (1) | |
Perfusion 1 | 150 mg/kg dans 200 mL* sur 1h |
Perfusion 2 | 50 mg/kg dans 500 mL*sur 4h |
Perfusion 3 | 100 mg/kg dans 1 00 mL* sur 16h |
* soluté glucosé à 5% ou chlorure de sodium à 0,9%
Les effets indésirables de la NAC sont le plus souvent non graves (urticaire, rash, troubles digestifs), mais le profil de sécurité inclut également des réactions d’hypersensibilité ou anaphylactoïdes plus graves. Ces dernières sont plus fréquentes et plus graves lorsque le médicament est perfusé trop rapidement ou en trop grande quantité (1). Au Canada où cette problématique a été plus précisément étudiée, certaines communications institutionnelles rapportent des conséquences graves voire fatales de certains surdosages et notamment dans des contextes d’erreur d’administration. Les manifestations rapportées incluent en particulier confusion, agitation, céphalées ainsi que des complications neurologiques plus sévères comme des crises convulsives, des œdèmes cérébraux pouvant évoluer vers un engagement cérébral ou encore des syndromes hémolytiques et urémiques. Des cas de décès ont ainsi été rapportés après des erreurs médicamenteuses suite au maintien du débit de perfusion 1 (dose de charge) (2,3).
Ce type d’évènement souligne la nécessité d’une vigilance accrue lors de l’utilisation de cet antidote. La sensibilisation des équipes au protocole antidotique recommandé et la sécurisation des étapes de prescription, préparation et administration de ce médicament constituent des leviers essentiels pour réduire le risque d’erreur et sécuriser la prise en charge des patients.
Références :
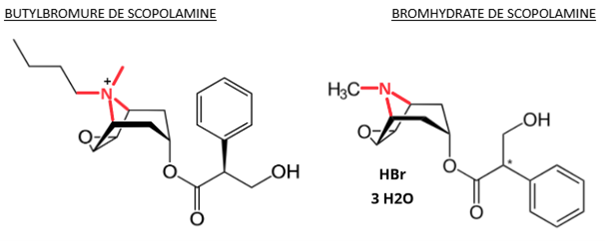
La pharmacochimie dans l’analyse des effets indésirables : un exemple d’erreur médicamenteuse avec la scopolamine
Christophe Benoit (interne) (Toulouse)
Notre Centre Régional de PharmacoVigilance a été interrogé sur une erreur d’administration chez un patient ayant reçu du bromhydrate de scopolamine au lieu de butylbromure de scopolamine par voie injectable.
Rapidement, le patient a présenté confusion, dysarthrie/aphasie, mydriase et xérostomie témoignant d’un effet atropinique. La scopolamine est un antagoniste muscarinique, utilisé dans les spasmes digestifs et urinaires, dans le mal du transport ou pour réduire les sécrétions bronchiques en contexte d’urgence ou de soins palliatifs. La scopolamine par voie injectable est conditionnée avec différents sels : bromhydrate de scopolamine et butylbromure de scopolamine.
Cette différence est liée à la localisation d’un atome d’azote : il a 4 liaisons covalentes avec des atomes de carbones lorsque la scopolamine est sous forme butylbromure, soit une fonction amine quaternaire, et 3 liaisons covalentes lorsque la scopolamine est sous forme bromhydrate, soit une fonction amine tertiaire. Cette différence de structure chimique se répercute sur le profil pharmacocinétique : la forme butylbromure, ne passe pas la barrière hématoencéphalique (BHE) alors que la forme bromhydrate, avec son amine tertiaire, passe la BHE.
De ce fait, les effets indésirables atropiniques au niveau du système nerveux central s’avèrent plus fréquents avec le bromhydrate de scopolamine traversant la BHE et restent plus rare avec le butylbromure de scopolamine.
Toux et dyspnée transitoires après injection intramusculaire de testostérone : penser aux micro-embolies huileuses pulmonaires
Pauline Schiro (Toulouse)
L’énanthate de testostérone (ANDROTARDYL) et l’undécanoate de testostérone (NEBIDO), indiqués notamment dans l’hypogonadisme masculin, se présentent sous forme de solutions huileuses (huile de ricin) nécessitant une administration exclusivement intramusculaire.
La survenue d’une toux, d’une dyspnée, parfois associées à une hyperhidrose, une douleur thoracique ou une syncope pendant ou immédiatement après l’injection, peut évoquer une micro-embolie pulmonaire huileuse (POME), un effet indésirable rare mais décrit. Les symptômes sont le plus souvent transitoires et spontanément réversibles, avec une prise en charge symptomatique si nécessaire, notamment par oxygénothérapie.
Le mécanisme repose sur le passage intravasculaire accidentel d’une faible quantité de solution huileuse, responsable d’une micro-embolie pulmonaire. Cet effet est donc lié aux modalités d’administration plutôt qu’au principe actif lui-même et ne contre-indique pas la poursuite du médicament. Cet effet indésirable est décrit comme bénin et transitoire dans la littérature. Cependant des cas graves ont été rapportés avec l’undécanoate de testostérone, nécessitant un passage aux urgences ou une hospitalisation (1).
Bien que généralement non grave, cet effet mérite d’être connu des patients et soignants. Il convient donc de rappeler certaines précautions visant à réduire ce risque : injection lente, administration dans le muscle glutéal et réalisation d’un test d’aspiration avant injection afin de limiter le risque d’administration intravasculaire accidentelle.
- Meyer RJ, Mann M. Pulmonary oil micro-embolism (POME) syndrome: a review and summary of a large case series. Curr Med Res Opin. 2015;31(4):837-841.
Sécuriser l’usage du méthotrexate en ville et en oncohématologie : enjeux actuels, MTX-HD, grossesse et place du VORAXAZE
Juliette Descoeur (Montpellier)
L’utilisation du méthotrexate en médecine de ville et dans les secteurs spécialisés d’oncologie-hématologie demeure un défi en raison de sa marge thérapeutique étroite, de ses interactions nombreuses et des risques associés à son élimination. En ambulatoire, les difficultés les plus fréquemment rapportées concernent la confusion entre schéma hebdomadaire et prise quotidienne, les interactions médicamenteuses et une surveillance biologique parfois insuffisante. Les recommandations actuelles rappellent l’importance d’un bilan pré-thérapeutique structuré, d’une évaluation de la fonction rénale et hépatique et d’une supplémentation adaptée en acide folinique pour limiter les erreurs de dosage et optimiser la tolérabilité (1).
En oncohématologie, l’administration de méthotrexate haute dose (MTX-HD), définie par des doses supérieures ou égales à 500 mg/m², expose les patients à un risque important de néphrotoxicité en cas de retard d’élimination. Les référentiels d’experts soulignent la nécessité d’une préparation incluant l’hyperhydratation alcaline, l’arrêt des médicaments diminuant la clairance rénale du méthotrexate, la surveillance rapprochée du pH urinaire, de la diurèse, de la fonction rénale et de la méthotrexatémie, ainsi que l’ajustement du sauvetage par acide folinique selon les concentrations résiduelles (2,3,4). L’efficacité du folinate diminue lorsque les concentrations de méthotrexate demeurent supérieures à 10 µmol/L pendant au moins 48 heures, ce qui justifie une intensification transitoire tout en respectant ses limites et impose d’anticiper une stratégie de sauvetage avancé si le retard d’élimination se confirme (5,6).
Dans ces situations, la glucarpidase (VORAXAZE), une carboxypeptidase recombinante capable d’inactiver rapidement le méthotrexate circulant, constitue une option thérapeutique essentielle. Elle est indiquée chez les patients, adultes ou enfants, présentant un retard d’élimination important ou un risque élevé de toxicité sévère. Son administration est recommandée idéalement dans les 48 à 60 heures suivant le début de la perfusion de MTX-HD, sur la base de seuils de méthotrexatémie tels que ≥ 30 µmol/L à H36, ≥ 10 µmol/L à H42 ou ≥ 5 µmol/L à H48 selon le schéma perfusionnel (6,7). L’acide folinique doit être administré à distance d’au moins deux heures afin d’éviter son inactivation par la glucarpidase ; l’épuration extrarénale peut être discutée au cas par cas mais reste d’efficacité limitée du fait de la redistribution tissulaire (7,2). Pour les perfusions courtes à très forte dose (par exemple 8-12 g/m² en ≤ 6 h), une interprétation précoce des concentrations (H12-H24) est utile pour anticiper la décision thérapeutique et l’organisation logistique, tout en conservant les seuils décisionnels à H36-H48 et la fenêtre optimale d’administration de la glucarpidase à 48-60 h (6).
Dans le cadre spécifique de la prise en charge des grossesses extra-utérines, la réglementation a été récemment renforcée. L’ANSM a actualisé en 2024 et 2025 le cadre de prescription compassionnelle pour intégrer les données européennes concernant la génotoxicité du méthotrexate et a étendu à six mois la durée de contraception nécessaire après exposition. Ces mises à jour répondent également à des cas d’administration injustifiée dans des contextes de grossesse de localisation indéterminée, justifiant un renforcement des exigences diagnostiques en imagerie et l’harmonisation des protocoles d’utilisation et de suivi des patientes (9,10,11).
L’ensemble de ces éléments souligne la nécessité d’une harmonisation des pratiques entre la ville et l’hôpital, de la diffusion large des référentiels actualisés et d’une vigilance accrue quant aux interactions et au suivi biologique. En oncohématologie, la maîtrise des protocoles MTX-HD et l’utilisation appropriée de la glucarpidase représentent des leviers majeurs pour réduire la morbidité liée aux retards d’élimination. En milieu ambulatoire comme en contexte de grossesse extra-utérine, la sécurisation du circuit du méthotrexate demeure un enjeu central pour limiter les risques iatrogènes et assurer une prise en charge optimale.
- RecoMédicales. Recommandations pour l’utilisation du méthotrexate. Bilan pré-thérapeutique, interactions, acide folinique. 2025. [academic.oup.com]
- Institut national du cancer. Prévention et gestion de la toxicité rénale induite par le méthotrexate haute dose. Avis d’experts, 2023. [ansm.sante.fr]
- ANSM. Méthotrexate haute dose : mesures générales de prévention du risque de néphrotoxicité. Actualisation 13/07/2023. [ema.europa.eu]
- Vidal. Méthotrexate haute dose : mesures de prévention contre la toxicité rénale. 2022, actualisé 2025. [has-sante.fr]
- HAS. Voraxaze (glucarpidase). Avis du 02/09/2022. [cancer.fr]
- Ramsey LB, Balis FM, O’Brien MM, et al. Consensus guideline for use of glucarpidase in patients with high-dose methotrexate induced AKI and delayed methotrexate clearance. The Oncologist. 2018;23(1):52-61. [vidal.fr]
- EMA. Voraxaze – Résumé des caractéristiques du produit. [kidneynews.org]
- OMEDIT Centre. Carboxypeptidase G2 – Voraxaze et surexposition au méthotrexate. Mise à jour 2022.
- ANSM. Décision du 27/06/2024 – Modification du CPC méthotrexate pour GEU (contraception 6 mois). [ashpublications.org]
- ANSM. Décision du 16/05/2025 – Modification du CPC méthotrexate (GEU) et renforcement du PUT-SP. [onclive.com]
- ANSM. Protocole d’utilisation thérapeutique et de suivi des patientes. 2025. [omedit-centre.fr]
Dosage du phosphatidylethanol dans le suivi de l’alcoolisme chronique
Léa Camail (Interne), Nicolas Fabre (PH), Anne Fontova (Ingénieure), Peggy Gandia (Toulouse)
La consommation chronique excessive d’alcool constitue un enjeu majeur de santé publique, étant impliquée dans près de 30 % des transplantations hépatiques réalisées chaque année en France (1).
Plusieurs biomarqueurs sanguins sont actuellement disponibles pour évaluer la consommation d’alcool, notamment les enzymes hépatiques, l’éthylglucuronide (EtG), l’éthylsulfate (EtS) et les phosphatidyléthanols (Figure 1). Parmi ces derniers, le dosage sanguin du phosphatidyléthanol 16:0/18:1 (POPEth),composé d’une chaîne d’acide gras saturé palmitique (16:0) et d’une chaîne d’acide gras mono-insaturé oléique (18:1), s’est progressivement imposé en pratique clinique comme un biomarqueur particulièrement sensible et spécifique de la consommation d’alcool. Il est aujourd’hui utilisé dans différentes situations cliniques, notamment pour le dépistage et le suivi de l’abstinence chez les patients présentant un trouble de l’usage de l’alcool, l’évaluation pré- et post-transplantation hépatique, en particulier dans les contextes de cirrhose, ainsi que dans certains contextes médico-légaux et de prévention des troubles du spectre de l’alcoolisation fœtale (2).
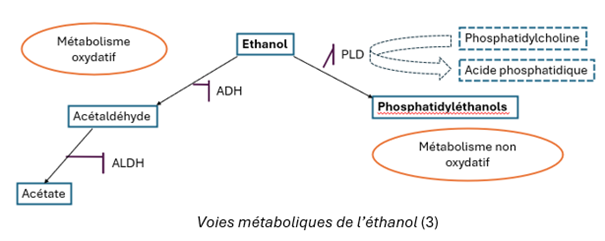
Plusieurs méthodes LC-MS/MS de dosage du POPEth ont été publiées, mais la majorité reposent sur des extractions en phase solide (SPE) ou liquide supportée (SLE), coûteuses en routine (4). À ce jour, ce dosage est réalisé uniquement par les laboratoires de Lille, Limoges et Nice (5), avec un délai de rendu d’environ 2-3 semaines (6).
Récemment, le laboratoire de Pharmacocinétique et Toxicologie du CHU de Toulouse a développé une méthode analytique LC-MS/MS en mode MRM reposant sur une extraction liquide/liquide (LLE). Cette méthode intègre un prétraitement par ultrasons, étape particulièrement déterminante pour l’extraction des phosphatidyléthanols associés aux membranes érythrocytaires (7,8).
Le dosage du POPEth constitue une alternative simple, rapide et moins coûteuse par rapport aux approches actuellement utilisées. Ce dosage permettra non seulement de réduire significativement les délais de rendu de résultats pour les demandes émises par les services au CHU de Toulouse, mais également de répondre à une forte demande des services cliniques de toute la région Occitanie.
1. La consommation d’alcool et ses conséquences en France en 2024 | OFDT [Internet]. 2025 [cited 2026 May 23]. Available from: www.ofdt.fr/publication/2025/la-consommation-d-alcool-et-ses-consequences-en-france-en-2024-2624
2. Schröck A, Thierauf-Emberger A, Schürch S, Weinmann W. Phosphatidylethanol (PEth) detected in blood for 3 to 12 days after single consumption of alcohol—a drinking study with 16 volunteers. Int J Legal Med. 2017 Jan 1;131(1):153–60. doi:10.1007/s00414-016-1445-x
3. Nguyen VL, Haber PS, Seth D. Applications and Challenges for the Use of Phosphatidylethanol Testing in Liver Disease Patients (Mini Review). Alcohol Clin Exp Res. 2018;42(2):238–43. doi:10.1111/acer.13558
4. Lahr RG, Sharma P, Maus A, Langman LJ, Jannetto PJ. Development of an LC-MS/MS assay with automated sample preparation for phosphatidylethanol (PEth)– Not your typical clinical marker. J Chromatogr B. 2023 Sep 1;1229:123886. doi:10.1016/j.jchromb.2023.123886
5. Annuaire et Catalogue des analyses - GT STP-PT [Internet]. [cited 2026 May 23]. Available from: sfpt-fr.org/catalogue-des-analyses
6. ubilab-result [Internet]. [cited 2026 May 23]. Available from: www.ubilab.io/software/ubilab-result
7.Perilli M, Toselli F, Franceschetto L, Cinquetti A, Ceretta A, Cecchetto G, et al. Phosphatidylethanol (PEth) in Blood as a Marker of Unhealthy Alcohol Use: A Systematic Review with Novel Molecular Insights. Int J Mol Sci. 2023 Jan;24(15):12175. doi:10.3390/ijms241512175
8. Yerra NV, Pini C, Cordon-Cardo C, Mendu DR. An improved and economical LLE-LC/MS method for quantifying phosphatidylethanol: Identification and separation of isotopic cross-talk in clinical alcohol testing. Clin Biochem. 2025 Oct 1;139:110994. doi:10.1016/j.clinbiochem.2025.110994

BIP Occitanie fait partie de l'International Society of Drug Bulletins (ISDB)